Article Text

Abstract
Background Gene therapies have the potential to be a curative approach to a large number of genetic diseases. However, granting of a positive marketing authorisation does not equal patient access to therapy.
Objectives The purpose of this paper is to identify a full set of hurdles potentially preventing patient access to gene therapies based on the available literature.
Methods A review of the literature using systematic approach in two distinct databases was performed by identifying relevant, peer-reviewed publications, between 2012 and 2018.
Results Seven major topics were identified as potential patient access hurdles, namely affordability, assessment of value, development of therapy, ethical/social factors, evidence generation, operational implementation and regulatory hurdles. From these, 25 additional subthemes were further identified. The most frequently mentioned obstacle in the literature is related to the affordability aspect especially focusing on high cost of therapy (84%) and therapy payment/reimbursement (51%). Importantly, the evidence generation focusing on limited trial outcomes (81%) seems as a strong obstacle in patient access to these therapies.
Conclusions A growing number of gene therapies are expected to be developed and made available to patients and healthcare professionals. Improvement of patient access to gene therapies can only be achieved by understanding all hurdles, in a complete and integrated fashion, so that strategies are timely established to ensure gene therapies’ benefits are provided to patients and to the society.
- accessible
- affordable
- economics
- global health
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Gene therapy medicinal products (GTMPs) are exceptionally promising treatment strategies, with the potential to cure a wide array of genetic diseases.1 Extensive research has been conducted in the field. According to Hanna et al, the number of clinical trials using GTMP as medicinal product has increased steadily over the past few years.2
This trend is aligned with the growing number of commercialised GTMPs worldwide. In Europe, the first GTMP approved was Glybera, in 2012. Since then, and until end of 2019, six additional products reached a positive marketing authorisation outcome (Imlygic, Strimvelis, Kymriah, Yescarta, Luxturna and Zynteglo). Conversely, in the USA, the first GTMP reached the market in 2017, while in the same time period a total of five approved products (Imlygic, Kymriah, Yescarta, Luxturna and Zolgensma) GTMPs are available.3 4
Development of GTMPs is a challenging process. Our previous research suggests that the main driver for negative marketing authorisation outcome in Europe is insufficient clinical efficacy evidence as well as safety issues, while issues at quality or non-clinical level play a secondary role in marketing authorisation application (MAA) outcome.5 Regulators are aware that advanced therapy medicinal products (ATMPs) aim at diseases of high unmet medical need. Therefore, in Europe, several strategies have been implemented to expedite the MAA process, such as the implementation of the innovative task force, the ATMP classification, the ATMP certification, the PRIority MEdicines scheme and scientific advice.1 5
However, a positive MAA outcome should not be considered an immediate synonym of therapy availability to patients. In Europe, after regulatory approval, health technologies are assessed for their value with subsequent (or parallel) pricing and reimbursement negotiations.6 In the case of gene therapy, this aspect is of particular importance considering the significant budget impact in health systems that GTMPs may elicit. Although high cost and budget impact are undoubtedly critical aspects in patient access to innovative therapies, additional factors should be taken into account.
This research intends to provide a comprehensive review of patient access to gene therapy by identifying a full set of hurdles. A review of the literature using systematic approach in two distinct databases was performed to identify relevant, peer-reviewed publications, between the years of 2012 and 2018. Data extraction was performed and qualitative synthesis is here presented.
Methodology
Search strategy
The Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines for conducting systematic reviews were followed. This study included publications available in Medline (accessed via PubMed) and Embase (accessed via Ovid) published between 1974 and 20 January 2019. The search strategy was purposefully designed to be broad, in order to ensure all relevant material was included.
Our search included both mapped and unmapped terms. Within the conducted search, “Boolean Operator” rules were used. The terms used were searched using ‘AND’ to combine the keywords listed and using ‘OR’ to remove search duplication where possible. Full search strategy is available as online supplemental appendix 1.
Supplemental material
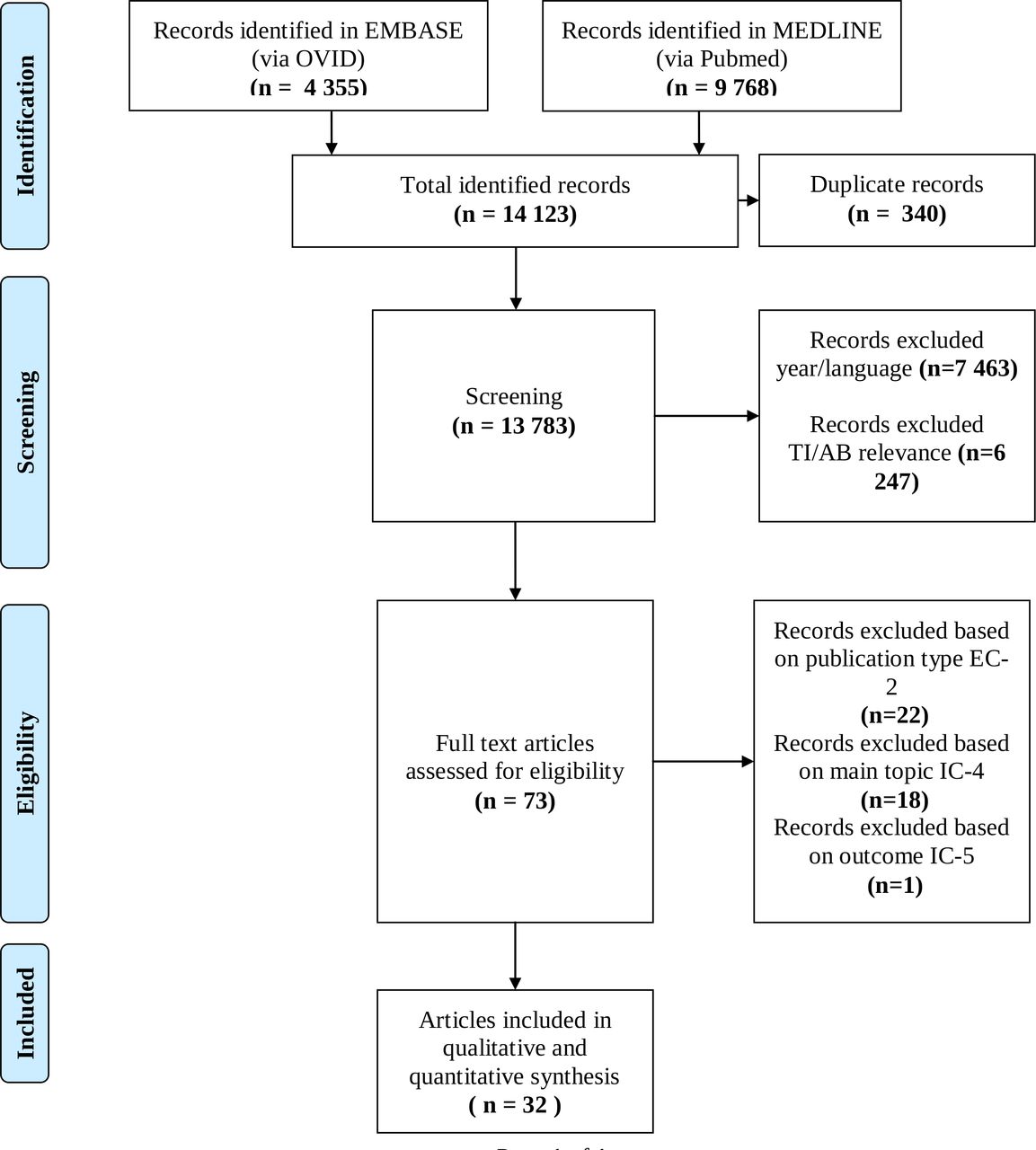
The process of identification, screening and inclusion of papers for this review is detailed in figure 1. Records were extracted to EndNote V.X8. The software deduplication functionality was used to identify duplicate references. Additionally, manual deduplication was performed. Following full text review, references were further excluded based on eligibility criteria described in table 1.
Study inclusion and exclusion criteria
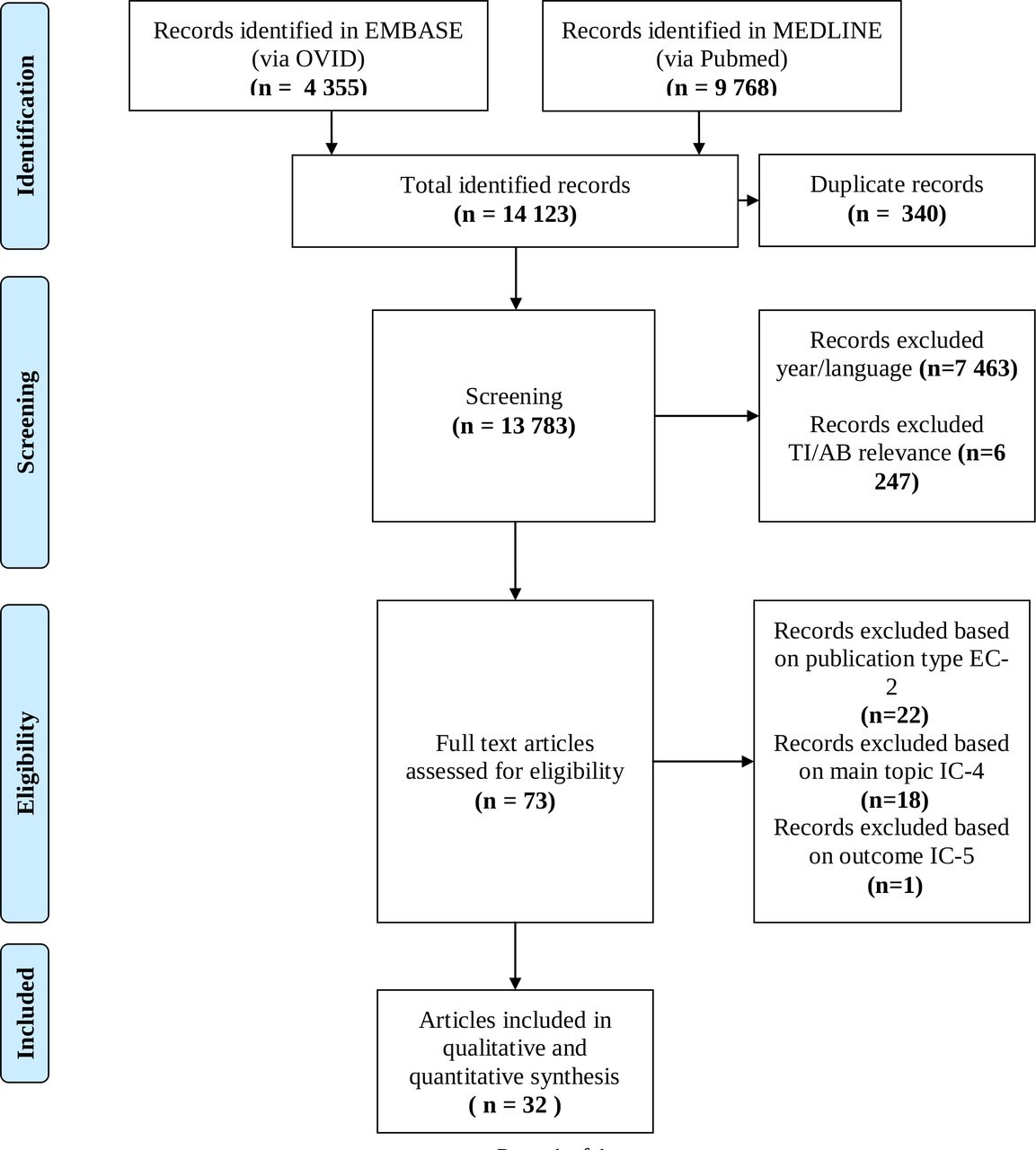
Process of identification, screening, eligibility and inclusion in this comprehensive review. TI/AB: Title/Abstract.
Publication selection
Eligibility criteria were developed in order to reflect the research aim. First, papers were included if they were published between 2012 and 2018. The year of 2012 was selected as lower cut-off date as it was the year that the first GTMP was approved in Europe. Only full-text articles published in peer review journals were included.
Articles which referred to the topic including but not limited to GTMP were included as well. This means that if a paper discussed cell and gene therapy simultaneously, this publication was included in the analysis. Additionally, at least one challenge related to patient access had to be extracted from the full-text review, in order to include the publication in the analysis.
Publications were excluded if not written in English. Other publication types such as books, book chapters, news articles/press release, and congress abstracts/posters were excluded. Eligibility criteria are fully detailed in table 1.
Data extraction and analysis
Publication characteristics were extracted from all relevant articles and were recorded in an extraction table. One researcher (MC) compared and extracted data and discussed any discrepancies with other researchers (BS, APM), when required. An overview of the identification process is documented in the PRISMA diagram, in figure 1.
Hurdles towards patient access were extracted from the full-text review of the articles. Major themes and subthemes were pulled from the data, until no more major topics and subtopics could be identified. Narrative synthesis of the articles was performed. A qualitative and quantitative analysis of the extracted hurdles is here presented.
Results
The search in both databases identified 14 123 publications. After removing 340 duplicates, a total of 7463 references were excluded based on year and language. Then, all remaining titles and abstracts were reviewed by MC for relevance, in alignment with the main objective of this research project, where 6247 were excluded. A total of 73 full text articles were reviewed. Studies were excluded if they did not meet the eligibility criteria specified in the study. Twenty-two records were excluded based on exclusion criteria 2 (publication type), 18 records were excluded based on inclusion criteria 4 (main topic) and 1 record was excluded based on inclusion criteria 5 (outcome).
In this analysis, 32 publications were included in qualitative and quantitative synthesis. These publications generated 7 major themes with 25 subthemes, which are described in table 2.
Major themes and subthemes extracted from the included literature
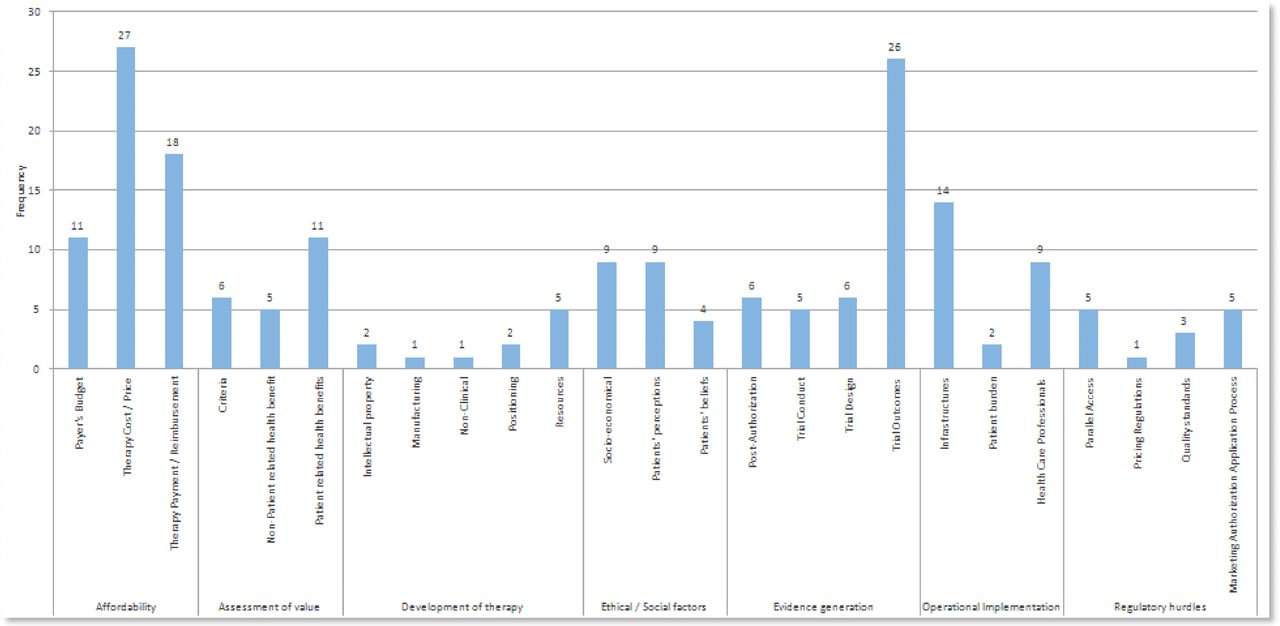
A frequency graph was generated which presents the number of publications out of the 32 which mention a specific hurdle, in each subtheme (figure 2).
{kind=link}
{kind=link}
Frequency of factors identified to influence patient access to gene therapy.
The six most common hurdles found in this comprehensive review belong to the themes/subthemes described below:
Twenty-seven publications (84%) mentioned affordability issues (therapy cost/price).
Twenty-six publications (81%) mentioned evidence generation issues (trial outcomes).
Eighteen publications (51%) mentioned affordability issues (therapy payment/reimbursement).
Fourteen publications (44%) mentioned operational implementation issues (infrastructures).
Eleven publications (34%) mentioned affordability issues (payer’s budget).
Eleven publications (34%) mentioned assessment of value issues (patient-related health benefits).
Each theme and subthemes are described in more detail in the discussion section.
Discussion
Affordability
Therapy cost/price
The price hurdle is not just related to the excessive high cost of these therapies.2 7–25 The price level is mainly justified by the high cost of development and manufacture.13 Some authors also mention that the increased medical follow-up associated with detecting late toxicities contributes to added costs.20 26 Furthermore, higher therapy cost is a possibility in special scenarios (eg, cost of gene therapy for patients with haemophilia who develop some level of immunogenicity, ie, antidrug antibodies is higher).22
On the other hand, many gene therapy products were developed using government/public funds. However, when setting up the final price, this is not taken into account.10 26 Estimating annual and lifetime costs is challenging due to variability in disease presentation, type, frequency of treatments required, access to follow-up care and payer source.21
Payer’s budget
Such an elevated price clearly has a significant impact on the healthcare budget impact.2 10 13 15–17 20 25 27 For example, in the USA, it is estimated that 25–30 million Americans have a rare condition related to a genetic defect. Considering the initial pricing experience with gene therapy in Europe, if more gene therapy products are made available at US$1–2 million price, the cumulative budget impact would be substantial and perhaps unsustainable. Assuming that gene therapies are developed to treat only one in 10 patients with a genetic condition, the cumulative budget impact at that price could rise to US$3 trillion, which is as much as the current spent in a year on all healthcare in the USA.28
Hurdles at the level of payer’s budget are even more marked considering the high pressure to control healthcare budget.13 20 This is especially true considering, for instance, in Europe, the period of recovery after a financial crisis and flattening of gross domestic product growth.2 In March 2017, a budget impact test was introduced in England, which assesses whether a new therapy’s aggregate additional cost to the healthcare budget exceeds the threshold value of £20 million per year. If the additional cost associated with the new therapy is expected to exceed this threshold in any of the first 3 years after launch, then additional commercial negotiations and potential restrictions apply.17
Therapy payment/reimbursement
Some authors mentioned the heterogeneity in reimbursement/payment strategies across different countries that geographically lead to different levels of access.10 27 It was clear from our research that standard reimbursement strategies may not be adequate to cope with super high cost treatments.10 13 14 21 Moreover, the reimbursement decision takes place after positive marketing authorisation, and this process may be lengthy in some cases.10 Even after the full assessment, health insurers may refuse to reimburse therapy. Some publications mentioned a lack of willingness to pay from governments and payers leading to no reimbursement after approval.19 22 26
An issue occurring particularly in the USA is related to the uncertainty on how to manage patients switching health plans. The first insurance company may be stuck with full upfront payment and no downstream benefit in case the patient decides to switch insurance company.10 12 21
With regards to discount rates, it is not clear how to reach the appropriate discounting rate. Gene therapies are likely to involve high intervention costs occurring years before all health effects have emerged.15
Finally, in case a payment based on performance is implemented, there is uncertainty on which outcomes to monitor. Hard endpoints are preferred, though this may not be possible in all cases.10
Assessment of value
Criteria
Heterogeneity in value assessment systems across different countries results in different coverage recommendations based on how health technology assessment (HTA) agencies perceive evidence and uncertainties. These different recommendations clearly lead to different levels of patient access to therapy. Additionally, several publications reported that gene therapy products may be assessed by HTA agencies using the same criteria and scrutiny than other classic therapies, which may be inadequate, considering the specificities of gene therapy. HTA systems appear to not be prepared to assess curative therapies.15 16 21 23 28 29
Non-patient-related health benefits
Some publications included in this analysis also highlighted that HTA models may not account for all relevant elements for assessment of value for health system and society. For instance, the ability of patients to go back to work, the work productivity or the impact on caregiver burden are often not considered for treatment reimbursement.15 21 22 27 28
Patient-related health benefits
A number of publications included in this analysis highlighted that endpoints that matter to patients (eg, quality of life) are not aligned with efficacy outcomes for reimbursement (eg, disease survival). There seems to be a strong mismatch between payers’ and regulator’s needs.10 14–16 21 23 26 This divergence may lead to a drug being approved by the health authorities for commercialisation but not reimbursed and, therefore, with a low level of market uptake.
Similarly to the issues noted at the level of non-patient-related health benefits, some publications reported that HTA models may not account for all relevant elements for assessment of value for patients.15 21 22 28 For instance, most gene therapies have the potential to be curative therapies. These may be valued more highly by society as opposed to non-curative therapies. Cure of a disease at a young age could help produce significant gains in many aspects such as omitting the cost of avoidable comorbidities, lifelong management of complications but also reducing the economic impact over individual patients and their caregivers/families (ie, work productivity) compared with treatments that bring marginal gains over many years. There is little evidence that such balance is currently being included in HTA.22 28
Development of therapy
Intellectual property
It is know that the academia plays a strong role in the development of gene therapy products. A survey conducted in 2016 reviewed ATMPs clinical trials in the EU reported that the majority of the sponsors (62%) were non-profit organisations, representing academia, hospitals and charities.30 GTMP development is often initiated at the academic level and then leveraged by larger pharmaceutical companies after commercial agreements are established.
The academia/industry partnerships influence remains uncertain, as the merger of academic intellectual freedom with big business focuses on value will surely create conflict.29 31 If these divergences remain unresolved or take too long to reach a solution, this may impact patient access to GTMPs.
Manufacturing
Another issue, particularly regarding gene therapy products based on genetic cell modification (eg, chimeric antigen receptor (CAR) or T-cell receptor), is related to the composition of the cell product. Uncertainties remain regarding the content of drug product/drug substance. On drug development, investigators question which subtypes of cells should be included. The selection of specific cell subtypes may increase even more the manufacturing costs, impacting patient access. The choice of cells is key for therapy success. Additionally, how to measure such success may pose challenges. Currently, high uncertainty exists regarding therapeutic success biomarkers. Without measurable efficacy biomarkers, the therapy will never reach the patients.31
Non-clinical
During gene therapy development process, animal testing is key for advancing to clinical trials. This is a challenging process and lack of appropriate non-clinical testing may have a therapy fail before even reaching to first-in-human studies. Understanding cross-species variability, particularly regarding viral vectors tropism and transduction efficiency, is critical for predicting clinical outcomes. Appropriate development of validated preclinical assays will be key to clinical experience.29
Positioning
External competitive landscape may have a significant impact on GTMP development with consequences to patient access. For instance, in the case of haemophilia, if the companies that bring GTMPs to market already have traditional haemophilia products within their portfolio, their incentive to offer gene therapy for a low price may be lacking because the new technology would disrupt their existing market.22
Another example is the ongoing innovation on regular monoclonal antibody therapy that can directly compete with antibody gene therapy. The classic therapy is a less costly alternative, with less administration burden and may potentially have higher efficacy.32
Resources
The resources level at the developer may also be a key factor influencing patient access. As previously mentioned, development of gene therapy often starts in non-profit organisations. In our research, some authors noted the lack of manufacturers experience2 33 as well as lack of preparedness from market access strategy and launch sequence,2 as key aspects impacting patient access. Additionally, low resources for translational research from academia and early clinical trials29 and lack of reimbursement after approval may lead to disincentive for manufacturers to develop breakthrough therapies.17
Ethical/social factors
Patients’ beliefs
Core individual values and beliefs may influence whether patients’ choose to have GTMP treatment or not. In two publications, it was noted that some patients may be unwilling to receive GTMPs, for instance, due to religious beliefs.34 35 Other patients may be intrinsically against germline genome manipulation, thereby refusing treatment.9 Finally, a study showed that one of the biggest fears about receiving gene therapy was that patients would not receive all the information. This apprehension is directly related to the assumption that gene therapy may alter features such as identity and personality.35
Patients’ perception
Patients’ perceptions on gene therapy may play a powerful role in the level of access to therapy. Several publications highlight a general lack of genetic literacy, not only from patients but also from caregivers,24 25 36 37 which in turn contributes to an inaccurate perception of gene therapy. In one publication, the potential irresponsible use of novel technologies and unrealistic expectation of cure (eg, in the case of HIV) were also noted as a barrier related to patients’ perception.8 Furthermore, the fear that genetic therapy will be used by those with means to improve intellect, physical abilities and longevity, thereby enhancing social inequality was also noted as a potential access barrier.11 35
Additionally, patients may be unwilling to receive genetic therapy due to psychological challenges (eg, receiving news about testing positive for a genetic marker of disease).34
Finally, a study showed that the degree of gene therapy acceptance by the public is directly related with the seriousness of the condition. If the disease is very serious, then patients will be more willing to accept GTMP treatment.35
Socioeconomical factors
Socioeconomical, cultural and geographical factors may potentially restrict access to gene therapy. Different price setup according to geographical regions will result in different GTMP availability. Consequently, others may become ‘treatment tourisms’. Finally, according to geographical region, there may be differences in standard of care therapy cost. These differences lead to different comparisons and conclusions on gene therapy reimbursement, which may cause discrepancies in patient access.8 10 11 22 24 25 27 34 38
Evidence generation
Trial design
A USA publication by Hampson et al reported the implementation of fully blinded, placebo controlled studies with specific GTMPs, in specific indications, would require unethical sham procedures13 (i.e. those GTMPs that require invasive methods of administration like Glybera, where the patient is administered with multiple intramuscular injections).
Furthermore, challenges at the level of comparator identification have been reported. Here, those therapies developed for diseases where there is no treatment are the most affected.13 15 21 In many cases, there is no other choice but to assess data resulting from single-arm, open label or even observational studies, which are known to be less robust for benefit–risk evidence generation.
Finally, finding easily measured patient-centred outcomes to assess efficacy was reported as an important hurdle related to clinical trial design. Trials evaluating gene therapy may rely on surrogate outcomes, as opposed to clinical outcomes. For instance, in oncology setting, the use of data from progression free survival as a surrogate endpoint rather than data from overall survival as a clinical endpoint allows implementation of shorter duration trials, contributing to a more expedited regulatory assessment of a marketing authorisation. On the other hand, other less known surrogate endpoints may be used and, in that case, these need to be developed and validated, with limited data and limited time.
Weighing up the benefits and risks of any medicine is a complex process, as it involves the evaluation of a large amount and diverse type of data. The actual benefits and risks of any medicine are determined based on the information that is available at a given point in time, which often involves a fair level of uncertainty.
In case surrogate endpoints are used, frequently there is considerable uncertainty because these may not allow capturing the combined benefit–risk profile of a technology and a surrogate may not translate to benefits for a clinical endpoint.13 15
Trial conduct
With regards to clinical trial conduct, four publications reported that getting patients diagnosed and recruited into clinical trials, as well as promoting adherence to medical follow-up, is an important hurdle. Patients seems to be inherently reluctant to share their data and participate in clinical translation.13 15 25 26 This may potentially be related to patient’s limited knowledge of GTMPs. A study on patient’s perspectives regarding gene therapy for sickle cell disease reported lack of knowledge of gene therapy from patients (eg, patients had fear of getting HIV if the vector was based on inactivated HIV virus) and a perception that treatment with gene therapy would be unacceptably unsafe (ie, negative effects of concomitant chemotherapy in sickle cell gene therapy, potential new onset of cancer due to gene therapy, potential infertility problems).39
Trial outcomes
On reviewing the data generated through pivotal clinical trials, some hurdles have been identified which could potentially be an obstacle to patient access. First, a strong uncertainty related to safety data, whether short, medium or long term has been reported.13 15 21 23 29 31 32 For instance, for CAR-T gene therapy product Yescarta, a number of patients experienced citokine release syndrome (CRS) and unexplained neurotoxicity. CRS symptoms ranged from fever and myalgias to life-threatening unstable hypotension and respiratory failure. While treatable for most cases, fatalities have been reported. On the other hand, the use of integrating vectors has an inherent potential genotoxicity risk, which is of particular importance following past reports that primary immunodeficiency children treated with retroviral vectors developed cancer.
Additionally, uncertain long-term efficacy of gene therapy products has been reported as a hurdle in patient access by several authors.13 16–18 25 27 29 32 On one hand, most of the clinical trials for candidate new GTMPs are conducted in a limited patient population (ie, rare diseases) where the main clinical efficacy endpoint is fairly new to the regulators and scientific community. These endpoints may not be the best choice, but this only becomes clear after some time, based on the evolution of knowledge around the disease. A great example of this was the European regulatory assessment of Glybera, where the initially assessed primary endpoint was triglyceride reduction but later it was noted that this surrogate endpoint was too variable from patient to patient and postprandial chylomicron reduction was used instead. On regulatory approval, long-term efficacy is extrapolated from pivotal clinical trials and when such uncertainty is raised at the pivotal trial level, it is even more difficult to predict effectiveness.
Moreover, the durability of clinical effect remains questionable. On one hand, this may be due to the unpredictability of transgene expression. Immunogenicity may limit a prolonged expression, which could potentially be related to a decreased clinical effect. On the other hand, tissue targeting refinement may be needed to improving transduction efficiency.32
Based on the way trials are designed and conducted (limited patient population, limited follow-up time, limited experience in primary clinical efficacy/safety endpoint analysis) it becomes clear that both efficacy and safety evidence at launch may be extremely immature. This may have an impact on limiting therapy access to patients.2 10 13–15 17 20 21 23 24
Overall, there is the need for improved understanding of the role of specific disease factors in gene therapy outcomes. As time goes by, more knowledge is built, leading to a better selection of patient population, biomarkers and endpoints.29
Post authorisation
After regulatory approval, post authorisation data are a mandatory requirement not only for safety but also for effectiveness new drug monitoring. Securing drug reimbursement is also often based on obtaining real world evidence. This is particularly important for GTMPs, where approval/reimbursement may be obtained with incredibly limited number of patients and open-label, uncontrolled clinical trials.15–17 27
In some instances, there is the need of implementing a patient registry. Several challenges related to this method of collecting postauthorisation data have been reported,10 40 including but not limited to:
Low number of patients.
Long term follow-up which may lead to low retention rate.
High administrative burden (eg, establishing site contract, local ethics committee approval, site staff training, etc).
High associated costs (eg, registry oversight, costs associated with multiple sites, database set-up, etc).
Limited data quality (eg, who is contributing to the registry, that is, only physicians? Patients? Family/caregivers?).
Limited resources (eg, regulatory agencies often approve gene therapy conditional to the implementation of a disease registry. From an industry perspective, sponsors prefer a registry based on drug use, while regulatory agencies favour a broader disease-based registry).
Data privacy issues (eg, in USA, if reimbursement is based on implementation of a patient registry, the legislation would need to change due to issues with privacy legislation).
Operational implementation
Infrastructures
Gene therapy manufacturing and quality control process are lengthy and complex (eg, difficulty in large-scale production of clinical-grade vectors). Besides not being readily available, certain GTMPs have generally short shelf-life, which may be particularly challenging in cases of urgent need of therapy (eg, acute diseases).7 15 29
Access to therapy may also be influenced by the need for adequate healthcare infrastructures regarding gene therapy manufacturing, administration and pre/postadministration medical monitoring.8–10 13 16 20 23 24 38 One publication reported that major health system changes are required before gene therapy can be fully implemented, highlighting the current limitations in information technology systems and limited support tools for clinical use of the information.37
Finally, many therapies in precision medicine, and especially gene therapy, need to have an appropriate validated companion diagnostic test approved by regulatory agencies, which the availability may differ from country to country.25
Patient burden
Generally, gene therapy administration involves a heavy patient burden.7 16 Patients need to be hospitalised to receive therapy. The hospitalisation may be for a variable period of time, since it may also include either preadministration preparation and/or postadministration medical monitoring. The majority of traditional drugs are self-administered by the patient, or even administered by a close caregiver, in the comfort of their home environment. For the case of gene therapy, due to its unique characteristics, administration in the hospital setting is likely to be the rule. Here, one should take the patient’s perspective where an additional itineration from patient’s home to a specific healthcare facility (in this case, hospital setting) could potentially be a hurdle for patient access, in many aspects, such as additional time spent or additional resources. Strimvelis is an example of gene therapy administered only at one site in Italy, meaning that patients have to travel to that specific clinical setting to receive treatment.
Healthcare professionals
As a unique and very distinctive therapeutic strategy, compared with classic treatments, gene therapy requires formal healthcare professional training (eg, with regards to safety and rescue therapy should any life-threatening toxicity occur).7 8 10 15 16 21 37 Also, physicians should be adequately trained to clearly explain patients and caregivers the benefits and risks25 of gene therapy.
Finally, a higher than usual administrative burden is expected for gene therapy,20 related to electronic patient medical records completion by healthcare professionals as well as other administrative documents (eg, health insurance forms). Overall, the specific training and higher administrative burden will likely result on an increase in human resource workload.
Regulatory hurdles
Marketing authorisation application process
Hurdles related to MAA process have been identified to contribute to different levels of patient access to gene therapies. First, there is a lack of regulatory harmonisation regarding ATMPs definition.33 In Europe, the definition of ATMP is included in regulation 1394/2007/EC. However, when a sponsor requests a classification from the European Medicines Agency (EMA), this it is not legally binding, and each member state may classify the same product differently.
Additionally, there is a lack of regulatory harmonisation towards MAA approval resulting in a different number of approved ATMPs across different geographical regions. For instance, up to 2017, in Europe there were 9 cell and gene therapy products approved through centralised procedure, while in the USA there were 17 products.41 From a patient access perspective, this may generate differences in accessing treatment according to geographical region. Legislative flexibility exists in different jurisdictions, specifically created to facilitate access to therapy for products not yet centrally authorised, although this means additional time and resources spent.
One publication specifically focusing on academic developers also reported the lack or limited interaction with regulators which decreases chances of a positive MAA.33 In Europe, several regulatory strategies are currently in place to support new ATMPs early in the development process, such as requesting for scientific advice/protocol assistance (SA/PA). A recent study conducted by Bravery et al 42 analysed the first 22 ATMP MAA submissions to the EMA suggests that requesting SA/PA does not seem to be decisive in terms of successfully obtaining MAA, since all sponsors requested it. Large pharmaceutical companies requested more SA/PA compared with small–medium enterprises, where academic developers are included. On one hand, the initiative to request SA/PA comes primarily from the applicant, as well as the content of the advice that is sought. It seems fundamental to ask the right questions, on the right timing. In addition, and although it may be unexpected, non-compliance with the regulator’s advice can be accepted in some cases. For instance, for Imlygic, advice was sought regarding the primary endpoint. While the EMA advised to use progression-free survival or overall survival, the applicant decided to use durable response rate, which was considered acceptable by the EMA, with proper justification.
Finally, two publications highlighted the lack of reimbursement after approval which may cause withdrawal of MAA.17 41 For instance, in Europe, the sponsor of Provenge (Sipoleucel-T) requested withdrawal of MAA in May 2015. The MAA of Glybera (Alipogene tiparvovec) expired in October 2017 and the sponsor chose not to review it due to commercial reasons.
Quality standards
Academic centres are important contributors to GTMP development. A study by Pearce et al 33 has shown several interesting barriers at the level of quality standards that may have an impact on patient access. First, even though it is considered an essential process, GMP manufacture adds significant costs and complexity to the production process. Second, there are unrealistic expectations of product qualification by some national health authorities in terms of manufacturing process. Lastly, in the EU, there is the statutory requirement for a qualified person (QP) for the release of investigational medicinal products. QP release of each batch when a single batch treats a single patient is prohibitively expensive and may even be logistically impossible in some cases.
Pricing regulations
While there are heterogeneous pricing regulations across different geographical areas, it is clear that GTMPs with elevated price will increase financial pressure on healthcare budget. Payers are less and less willing to pay for therapy with immature evidence. However, in diseases of high unmet medical need, society is likely to exert pressure on politicians to get access to therapies. In this context, pricing regulations need to be reconsidered, taking into account the growing number of high-cost gene therapy products approaching the market.2
Parallel access
In Europe, hospital exemption (HE) is an alternative pathway to centralised marketing authorisation. HE is a permission that can be granted by EU member states for unauthorised ATMPs to be used on a named-patient basis in a hospital setting within the same member state only and under the exclusive responsibility of the treating physician. While theoretically this should promote patient access to GTMPs, the less stringent requirements in HE may put public health at risk.26 33 41 For instance, HE has been criticised because its implementation varies between member states, which has been said to put patients at risk (eg, due to nonroutine processing in small batches). In addition, a successful pharmaceutical industry’s lobbying resulted in attaining such a level playing field (ie, comparable competition environment/setting), in which the conditions for applying HE are kept as narrow as possible. Consequently, hospitals have more difficulty in competing with commercial actors manufacturing ATMPs. This has resulted in some valuable established therapies risking to become unavailable for patients in need of them.41 43 Additionally, the abusive use of parallel access pathways may result in withdrawal of centralised marketing authorisation.33 41
Current GTMP patient access landscape in Europe and USA
Other authors have attempted to review GTMP reimbursement status and HTA decisions in major European countries and USA, allowing us to reflect on gene therapy patient access in each individual country.44 45 In England, two GTMPs were reimbursed (Imlygic, Strimvelis) with patient access schemes. In addition, two CAR-Ts (Yescarta and Kymriah) were funded through the Cancer Drugs Fund.44 45 In Scotland, Kymriah was accepted for B-cell acute lymphoblastic leukaemia treatment with a patient access scheme, while Yescarta and Kymriah for diffuse large B-cell lymphoma were rejected due to unjustified cost-effectiveness estimates.44 In Germany, three GTMPs had ‘non-quantifiable added benefit’ due to insufficient data (Glybera, Yescarta and Kymriah) and Imlygic had ‘no-added benefit’ due to inappropriate comparator use. However, this did not limit its reimbursement.44 45 Three GTMPs were reimbursed in France (Yescarta, Kymriah and Luxturna), while Glybera was not recommended, as it was considered to have ‘insufficient’ benefit due to its unsustainable and heterogeneous treatment effects.44 45 In Italy, one GTMP was reimbursed for hospital use with managed entry agreement (Strimvelis).45 In Spain, Kymriah was recommended for use inspecialised centres.45 In the USA, Kymriah, Yescarta, Luxturna and Zolgensma were evaluated as having substantial net health benefits. However, a high certainty of conclusion for the assessment of Zolgensma was established44. No data on Zynteglo were available for any of the EU5 countries either because the assessment is in progress or not assessed at all.45
Overall, discrepancies among HTA bodies’ perception of GTMPs’ value were noticed. Hanna et al highlight that uncertainty due to lack of robust and long-term evidence was the main limitation in securing reimbursement. On the other hand, conditional reimbursement is increasingly considered a useful strategy to mitigate uncertainty as it allows collection of long-term data whist minimising the impact on patient access. Qiu et al refer that although the limitations in pivotal studies resulted in substantial uncertainties regarding long-term treatment benefit, there was still a possibility for gene therapies to gain acceptance from HTA bodies. Most importantly, further evidence collection becomes the critical key, not only to reduce the uncertainty in reimbursement decisions, but also to increase the public’s confidence in the use of gene therapies.
Both these studies results are aligned with the present study with special focus on the fact that evidence generation (trial outcomes) and affordability (reimbursement issues) are two of the most relevant hurdles in GTMP patient access. Qiu et al also highlight that value appreciation constitutes an important factor for patient access impacting reimbursement, since different countries showed different perspectives on the weights allocated to each attribute. In our study, the criteria used on value assessment were found in 6 out of 32 publications (19%).
Neither HTAs nor payers are a homogeneous group of decision makers. Different methodologies and factors have an impact on the decision process depending on these methodological differences, thereby influencing patient access to medicines. Characteristics of different payer types are available in table 3. Additionally, HTA agencies may be grouped in three key archetypes.46 Agencies, such as the German IQWiG/G-BA, base their decision on the clinical benefit assessment, while cost-effectiveness analysis is only conducted in case of disagreements during pricing negotiation. Others including the English National Institute for Clinical Excellence (NICE) and the Scottish Medicines Consortium (SMC), mainly base their decisions on cost-effectiveness analysis using the incremental cost-effectiveness ratio. Some ‘mixed’ HTA frameworks combine both clinical benefit assessment and health economic assessment when making their decisions (cost-effectiveness and budget impact analysis). Here, decisions are mainly driven by budget impact analysis rather than cost-effectiveness analysis. For instance, in Italy and Spain, cost-effectiveness analysis and budget impact analysis are not mandatory for national pricing and reimbursement application, but they can be submitted by the manufacturers.47
Key payer types
Conclusion
A limited number of GTMPs have successfully been granted successful marketing authorisation, in Europe. We have previously conducted a retrospective study focusing on hurdles that GTMPs face during the MAA process. Clinical efficacy and safety issues appeared to have a major impact in unsuccessful MAA outcome for GTMPs.5 However, a positive MAA does not necessarily mean that the therapy is actually being used by patients and healthcare professionals.
From this comprehensive review, seven major themes were identified as potential patient access hurdles towards gene therapy clinical application and 25 subthemes were further identified. The major themes are outlined below:
Affordability.
Assessment of value.
Development of therapy.
Ethical/social factors.
Evidence generation.
Operational implementation.
Regulatory hurdles.
Affordability issues especially related to therapy cost/price (84%) but also to therapy payment/reimbursement (51%) are those most mentioned throughout the publications included in this analysis. There is no question that providing a potentially curative therapy comes at a certain price, most of the times unprecedentedly high. Throughout the years, this has not been the case for traditional medicines, as often the new products are intended to treat rather than cure diseases. Overall pressure to control healthcare budget is elevated. The assessment of value provides a link between therapy benefits for the patient and for the healthcare system and the willingness to pay. The payment/reimbursement decision-making process is based on the generated evidence which often included fairly limited, not only in patient numbers but also in the follow-up time, as most gene therapy products target rare diseases. This uncertainty contributes to different levels of access to gene therapy, since with the same data have been noted that one product is reimbursed in one country but not in another, due to different criteria. Our research results seem aligned with other author45 48 in the sense that evidence generation (trial outcomes) and affordability (reimbursement issues) present two of the most relevant hurdles in GTMP patient access. Additionally, value appreciation is noted as an important hurdle for patient access impacting reimbursement. HTAs and payers are heterogeneous group of decision makers across jurisdictions and diverse assessment methodologies have an impact on the decision process.
The lack of relevant information (comparative data vs potential comparators, robust Quality of Life (QoL) data, collection of relevant outcomes, short trial durations) raises high uncertainty regarding the long-term efficacy and safety for most gene therapies. HTA bodies use different methodologies to minimise this uncertainty while accepting high cost GTMPs. Despite this, to date, most gene therapies have successfully been granted reimbursement, with more or less delay in terms of assessment timelines, as described elsewhere.44
In less extent, ethical and social aspects related to the use of genetic therapy also seem to impact patient access. It became clear that the more serious a medical condition is, the more likely the patient is willing to use gene therapy. Operational implementation of gene therapy also rises as an important access aspect, especially related to the need of having specific infrastructures for administration of therapy and medical follow-up, as well as trained healthcare professionals. Some hurdles (eg, patient perception, beliefs. etc) are applicable to all patients, regardless of geography.
Society and healthcare systems must adjust to this new reality. It is expected in the near future that more and more GTMPs are developed and made available to patients and healthcare professionals. Improvement of patient access and GTMP availability can only be achieved by understanding all hurdles, in a complete and integrated fashion. It is important to have these hurdles present so that clear strategies are set to overcome them since the significant benefits of gene therapy will not be realised unless patients have access to it.
Acknowledgments
MC would like to thank PRA Health Sciences and National Association of Pharmacies (ANF) for the support provided in this project.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors All authors planned and conducted the work described in the article. MC reported the findings. MC is responsible for the overall content as guarantor.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplemental information.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.